
D35.2 Acid Strength and Molecular Structure
We know that an equilibrium favors the thermodynamically lower energy (more stable) side of the reaction, and that the magnitude of the equilibrium constant reflects the energy difference (ΔrG°) between the reactants and products. Consequently, for an acid-base reaction equilibrium involving a generic acid (HA):
Anything that lowers the energy of A– relative to HA, or raises the energy of HA relative to A–, will make ΔrG° more negative, increasing Ka, and hence make HA a stronger acid (green trace in figure below). Similarly, anything that lowers the energy of HA relative to A–, or raises the energy of A– relative to HA, will make ΔrG° more positive, decreasing Ka, and hence make HA a weaker acid (red trace in the figure below).
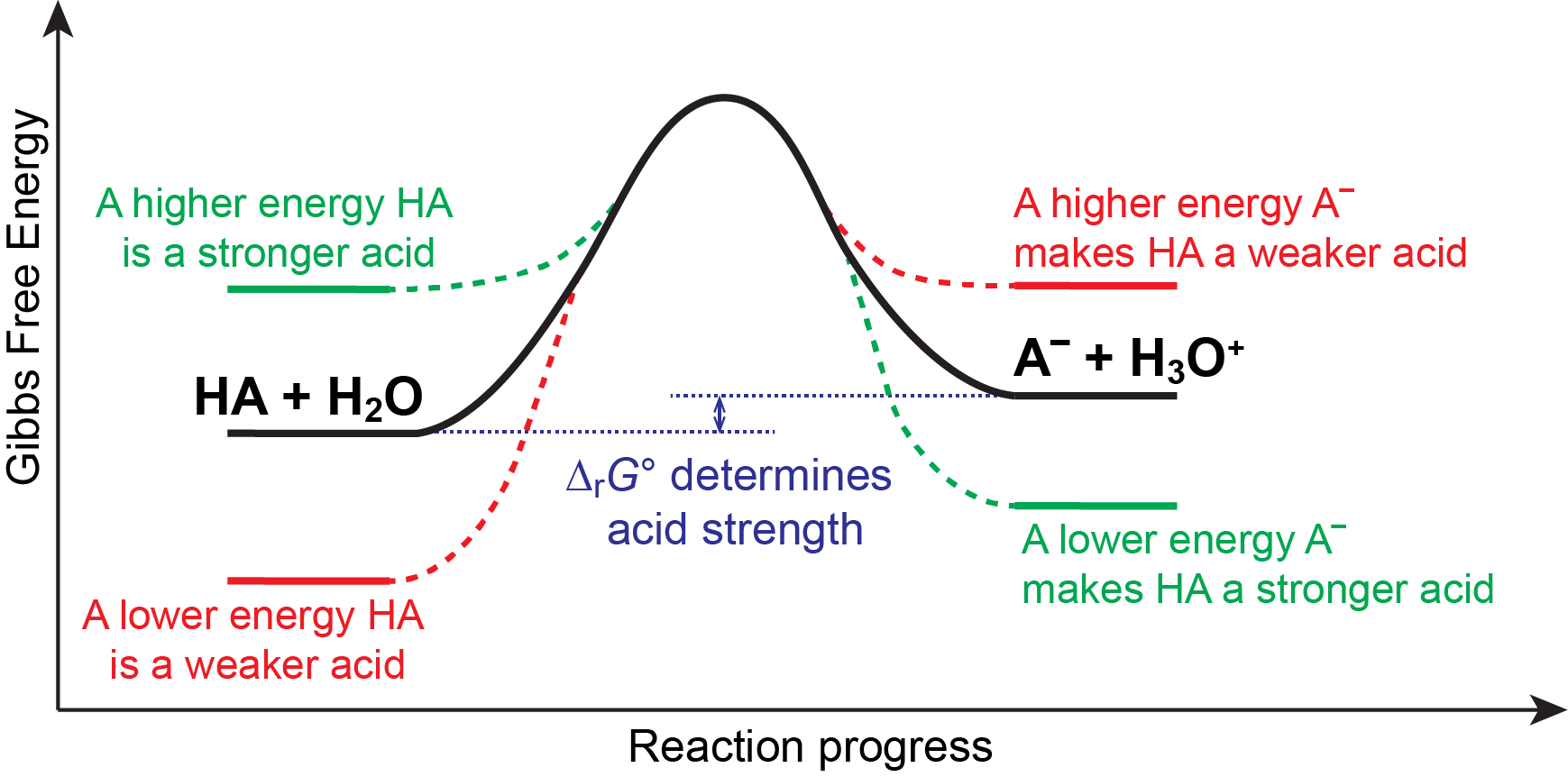
In other words, for a strong acid (Ka > 1), HA + H2O is higher in energy than A¯ + H3O+. This instability of the acid relative to its conjugate base drives its high reactivity, making it a strong acid. Similarly, for a weak acid (Ka < 1), HA + H2O is lower in energy than A¯ + H3O+, making it less reactive in an acid-base reaction.
Relative Acid Strengths
The strength of an acid, i.e., its Ka or pKa, is fundamentally a thermodynamic property, dictated by the ΔrG° of the reaction. As we compare the acidity of one acid to another, we are fundamentally comparing how the ΔrH° and ΔrS° of the reaction vary from acid to acid.
As an example, let’s consider the hydrogen halides. These acidic species are diatomic molecules composed of an H atom and a halogen atom.
| ΔrH° (kJ/mol) | ΔrS° (J/mol·K) | Ka (25 °C) | pKa (25 °C) |
|
| HF(aq) + H2O(ℓ) ⇌ F¯(aq) + H3O+(aq) | -16 | -102 | 6.8 × 10-4 | 3.17 |
| HCl(aq) + H2O(ℓ) ⇌ Cl¯(aq) + H3O+(aq) | -57 | -35 | 1.2 × 106 | -6.1 |
| HBr(aq) + H2O(ℓ) ⇌ Br¯(aq) + H3O+(aq) | -65 | -13 | 1010 | -10 |
| HI(aq) + H2O(ℓ) ⇌ I¯(aq) + H3O+(aq) | -62 | 11 | 1011 | -11 |
For HF, the reaction is overall exothermic (negative ΔrH°) due to a combination of bond breaking, bond forming, and changes in solute-solvent interactions:
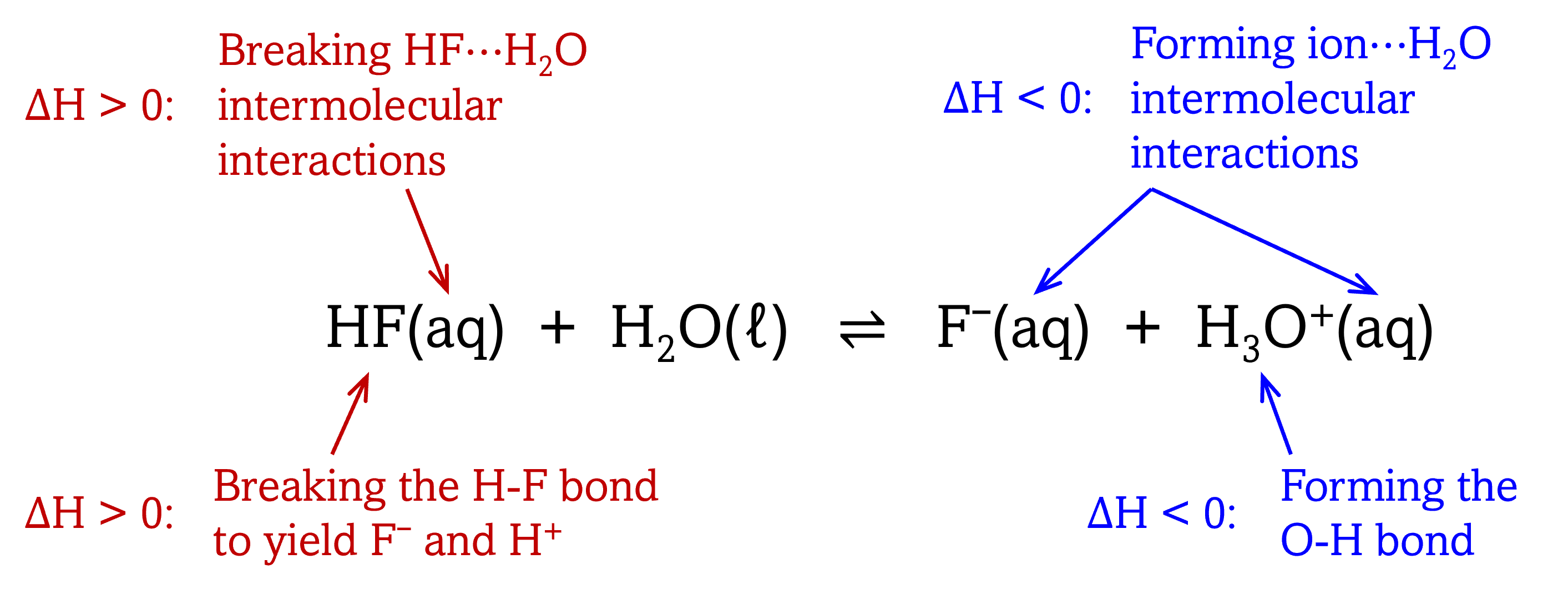
The entropy change for this reaction (ΔrS°) is also negative: the stronger ion-water interactions present in F¯(aq) and H3O+(aq) result in more restricted motions for the water molecules surrounding the ions compared to water molecules around the neutral HF. In other words, the solvated ions form a relatively rigid shell of water molecules around it, and just as H+(aq) can be thought of as H13O6+(aq), F¯(aq) can be thought of as F¯(H2O)n(aq).
Together, the ΔrH° and ΔrS° values leads to an overall positive ΔrG° at 25 °C and HF is a weak acid at room temperature.
We can see from the table above that Ka increases going down the halogen group, and HCl, HBr, and HI are all strong acids at 25 °C, with HI being the strongest acid in this series. While it may appear that this trend can be reasoned by comparing H-X bond strengths, which decreases with increasing X valence orbital size (due to worsening overlap with the H 1s orbital), looking closer at the ΔrH° and ΔrS° values shows that bond strength is only one part of the picture.
Left-click here for further comparisons of ΔrH° and ΔrS° values in hydrogen halides
Comparing the four reaction equations in the table above, we can see that the HX acids differ from each other in the H-X bond being broken and X¯ ion being formed; the formation of the O-H bond in H3O+ is the same in all four acids. Hence comparing relative bond strengths is one component in understanding the acid strength differences. (Note, the average bond enthalpy values are associated with a different bond breaking reaction than acid-base reactions, but they can still be used to compare bond strengths.) Comparing the electronegativity difference between the halides is another aspect (see section below).
Yet another component is the solvent interactions, both for the disrupted interactions in HX(aq) and the formed interactions in X¯(aq). Here, HF differs from the other three HX acids in that it is capable of forming strong hydrogen-bonds with water molecules.
However, of the four halides, F¯ has the strongest solvent interaction. So, the HF reaction is still exothermic. Moreover, ΔrS° is most negative for the HF reaction due to the strong F¯···water interactions: F¯ will structure water molecules around it to a greater extent than Cl¯, Br¯, and I¯.
Taken all together, as we go down the halide group, we see a generally more negative ΔrH° (although the differences diminish and appear to reverse), and a less negative ΔrS°. Combined, they give rise to the observed overall Ka trend.
In order to compare acid strengths quantitatively, we must consider both the enthalpy and entropy changes. This is when a reference table with thermodynamic values must be consulted.
Then how do we compare acid strengths in a more qualitative manner, if all we have on hand are our hand-drawn 3-D Lewis structures? We can begin by connecting molecular structures to relative energies, and consider factors that stabilize/destabilize the reactant and/or product (consider again the general ideas behind Figure: thermodynamics of acid-base reactions.)
Below, we will consider specific factors that would stabilize the presence of excess electron density. For an acid molecule that is itself neutral, which is the case for most acids, its conjugate base is negatively charged. Therefore, factors that can stabilize this excess charge can stabilize the conjugate base, thus increase the strength of the acid. (In this simplification, we approximate that the stabilities of the neutral acid molecules are comparable enough, such that they can be ignored in understanding the trends in acid strength.)
Electronegativity of the atom where the excess electron resides
Generally speaking, an excess electron would be at a lower energy when it resides on a more electronegative atom. For an example, let’s compare the relative acidity of the following second row molecules:
| Ka (25 °C) | pKa (25 °C) |
|
| CH4(aq) + H2O(ℓ) ⇌ CH3¯(aq) + H3O+(aq) | 1 × 10−50 | 50 |
| NH3(aq) + H2O(ℓ) ⇌ NH2¯(aq) + H3O+(aq) | 1 × 10-34 | 34 |
| H2O(aq) + H2O(ℓ) ⇌ OH¯(aq) + H3O+(aq) | 1.0 × 10-14 | 14 |
| HF(aq) + H2O(ℓ) ⇌ F¯(aq) + H3O+(aq) | 6.8 × 10−4 | 3.17 |
Here, the excess electron in the conjugate base resides mostly on the non-hydrogen atom, where the electronegativity trend is F > O > N > C. Therefore, F– can stabilize the extra electron better than OH–, which is in turn better than NH2–, which is better than CH3–. Consequently, HF can dissociate (forming H+ and F–) to a greater extent than H2O, and so forth.
If we place the reactant-side of these four acid-base reactions at the same energy, as in the figure below, the effect of the conjugate base stability on the acid strength is highlighted.
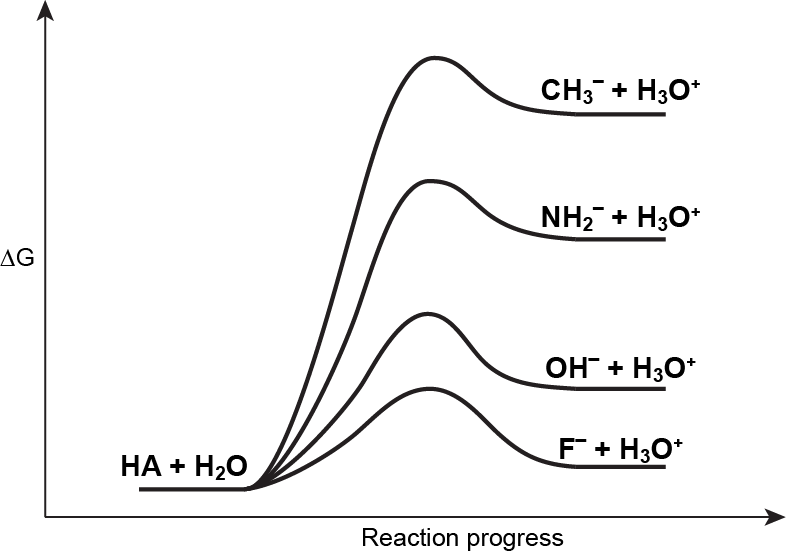
Electronegativity also influences the polarity of the A-H bond: as electronegativity of the non-H atom increases, the A–H bond becomes more polar, with greater δ+ on H, thus increasing the propensity for dissociation to form A¯ and H+.
Electron delocalization
The carboxylic acid functional group (-COOH) and alcohol functional group (-OH) both contains an O-H bond. When they act as an acid in an acid-base reaction, this O-H bond is broken, yielding an excess electron on the O atom:
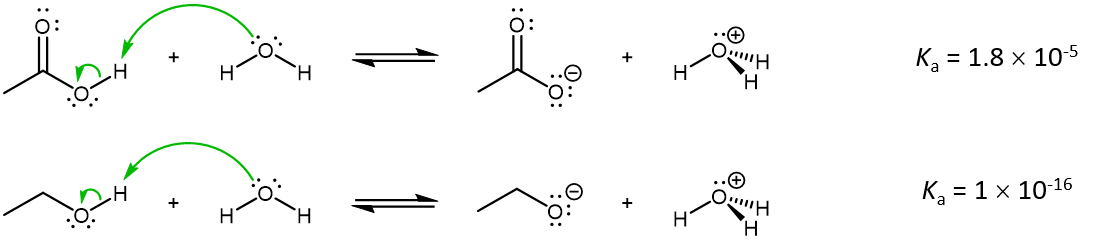
At a quick glance, given the similarities in these two reactions, it may be surprising that acetic acid is 10,000,000,000 times more acidic than ethanol. The reason again lies in the stabilization of the excess electron density in the conjugate base.
For the conjugate base of alcohols, the extra electron is localized on the single O atom. Hence, the acidity of ethanol is similar to that of water.
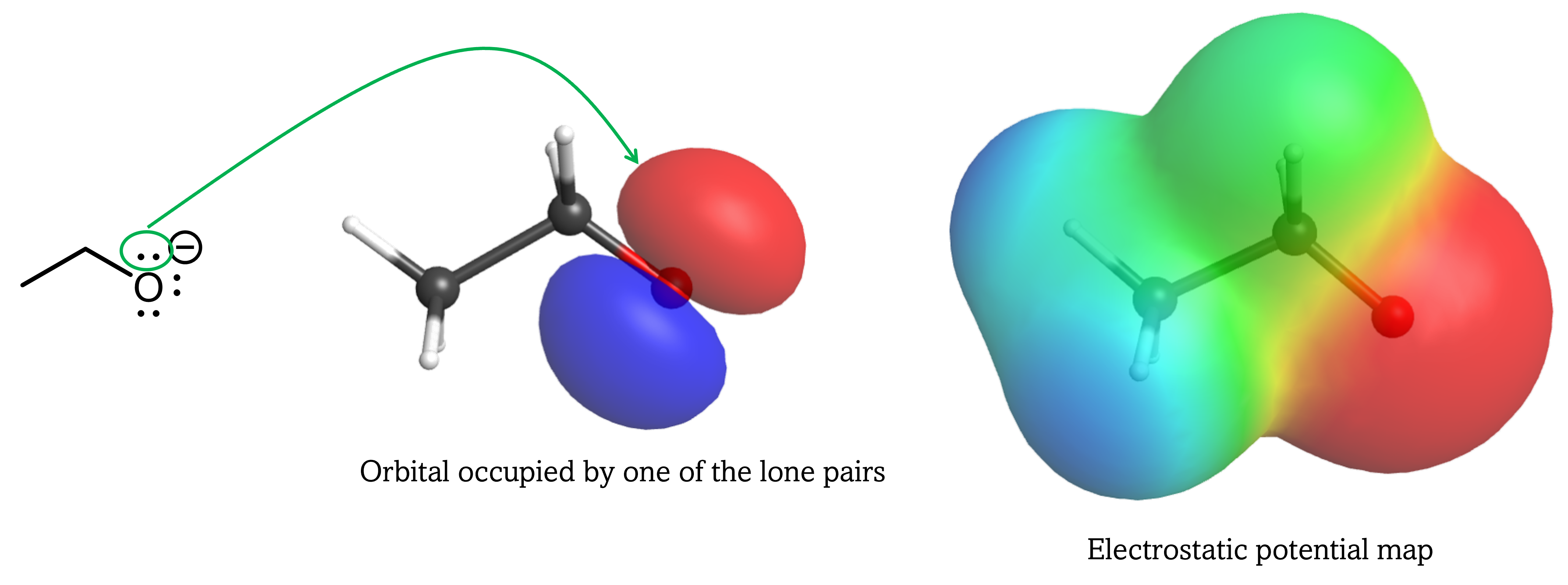
In contrast, the conjugate base of carboxylic acid has the extra electron delocalized over both O atoms, which necessitates drawing a set of resonance structures to properly describe the anion.
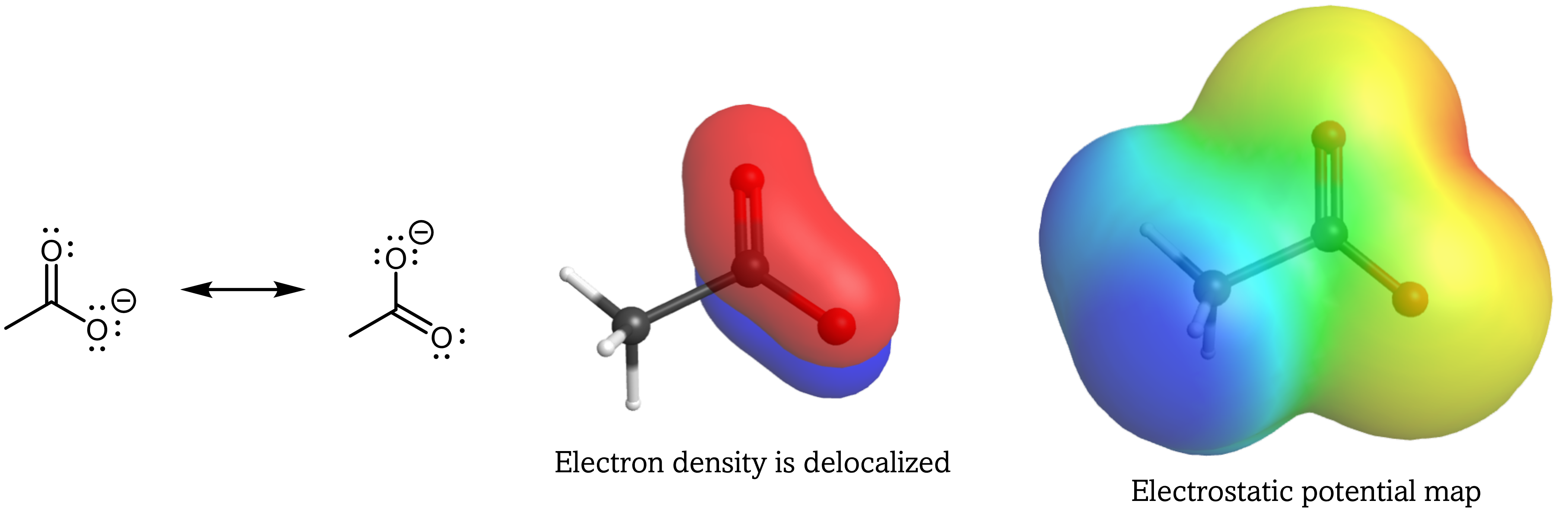
The two major resonance structures have equal contributions in describing the acetate anion, and the excess electron is effectively spread out over both O atoms. This delocalization of electron density, described by a set of resonance structures, stabilizes the conjugate base, which leads to a markedly increased acidity in carboxylic acids compared to alcohols.
Inductive Effect
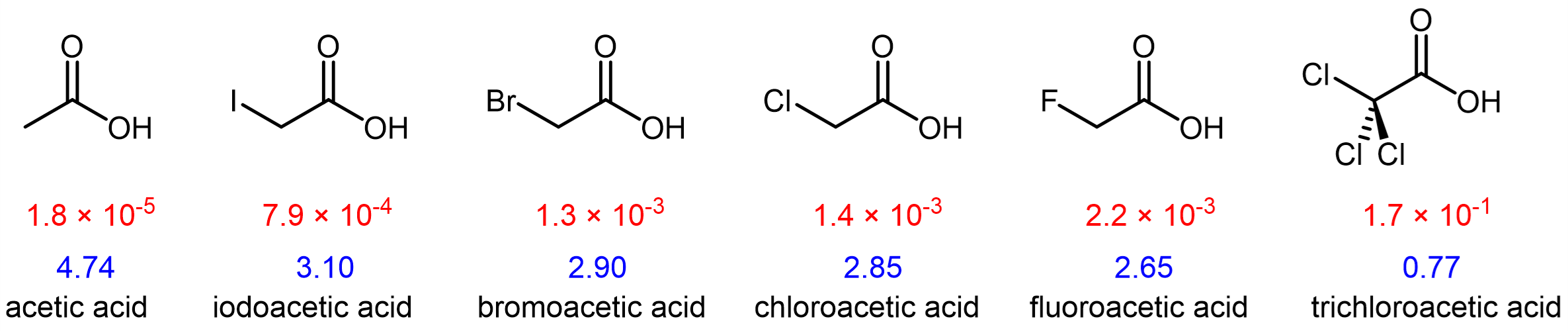
Atoms (or functional groups) in a molecule that are not directly bonded to (or contain) the acidic H can influence acidity via the inductive effect, that is, they can induce a polarization in the distribution of electrons within the molecule. For example, a more electronegative substituent (I, Br, Cl, or F) near the -COOH group can increase the acidity of the carboxylic acid.
This inductive effect works in two ways. First, it stabilizes the anionic conjugate base by drawing electron density away from the negatively charged -COO– group. It allows the excess electron to delocalize over multiple atoms, similar to the π-conjugation delocalization discussed above, via the polarity of the covalent bonds.
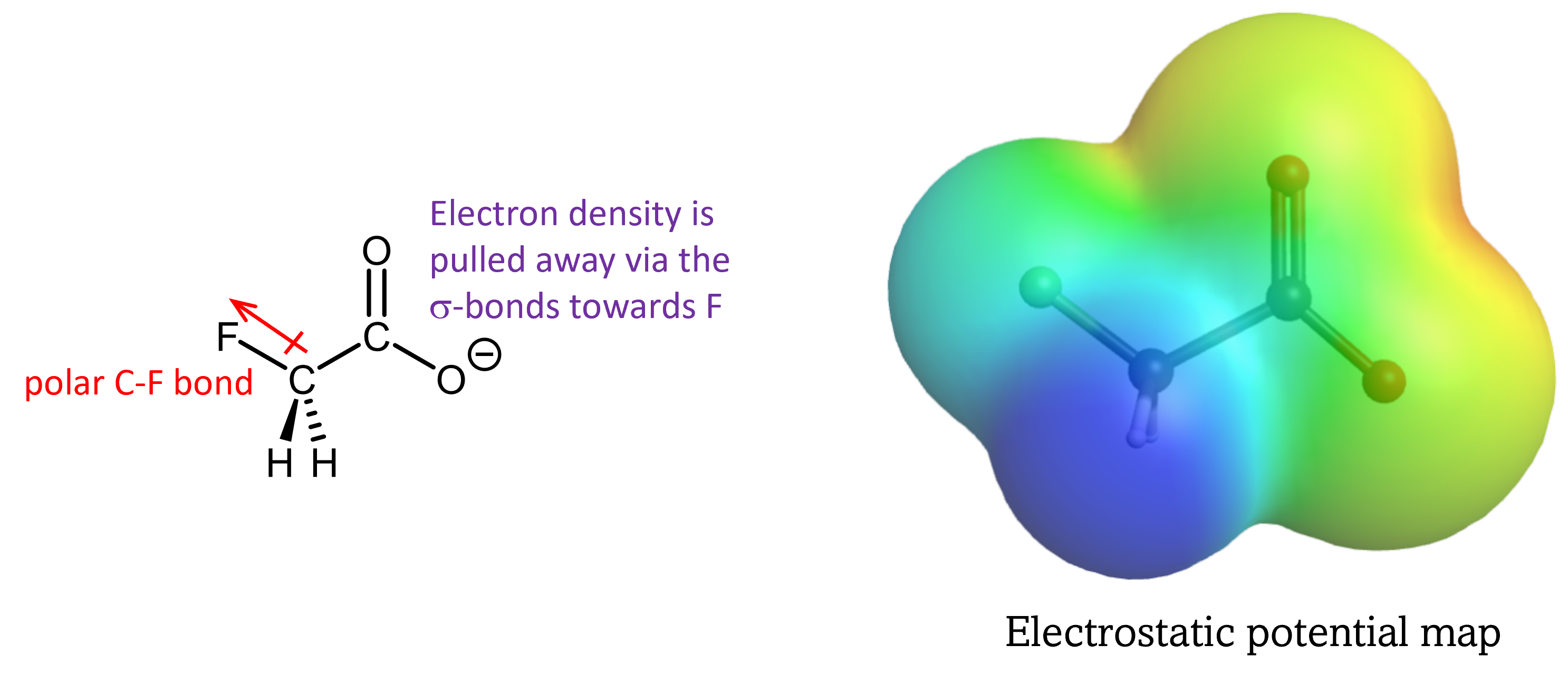
It also increases the polarity of the O-H bond in -COOH by drawing electron density away from this bond in the acid molecule, facilitating bond breaking and formation of H+.
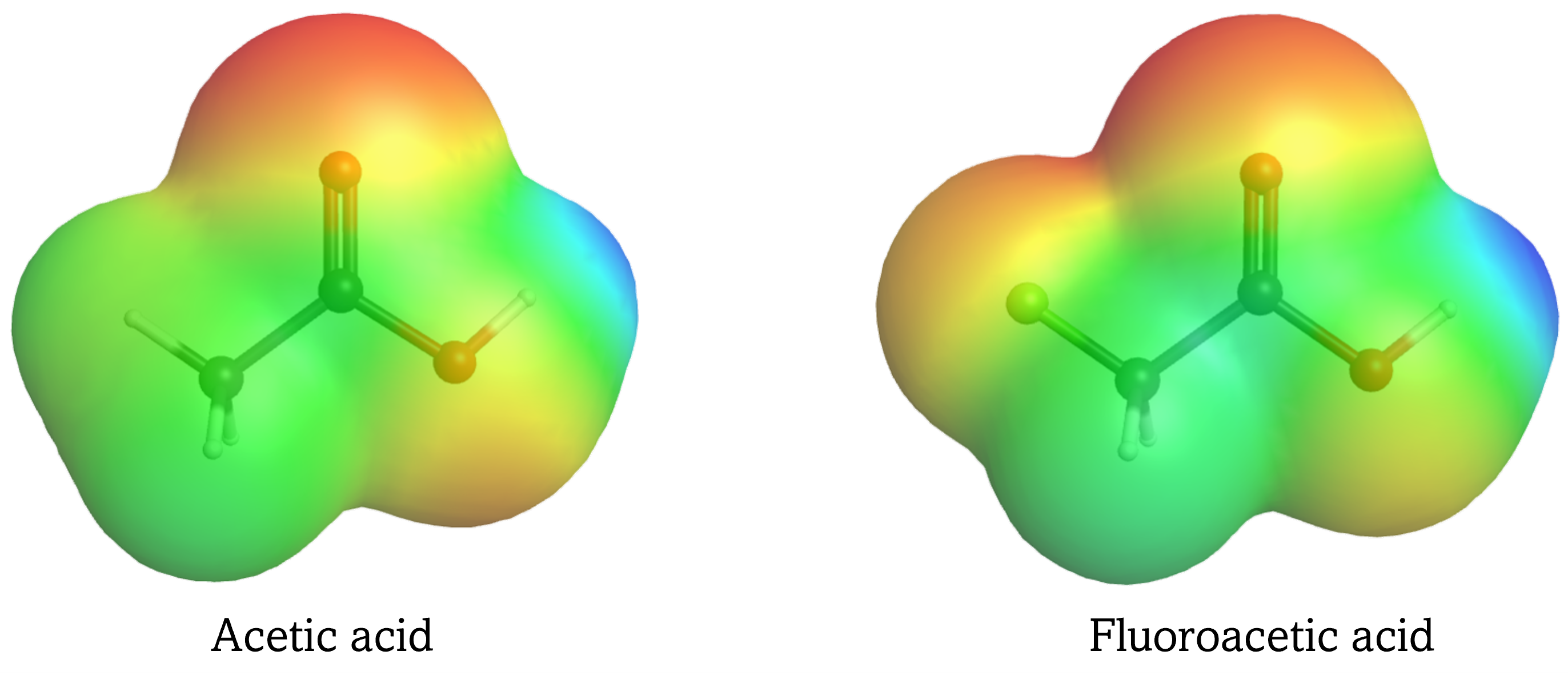
The magnitude of the inductive effect depends on the nature as well as the number of substituents. For example, F, which is more electronegative than Cl, causes a larger inductive effect as it draws greater amount of electron density to itself. Having three Cl atoms, on the other hand, leads to an overall larger inductive effect, making trichloroacetic acid more acidic than fluoroacetic acid.
Activity: Inductive Effect 1
The nature of substituents would also affect the direction of the inductive effect. In other words, instead of drawing electron density away from the -COO¯ group like the halides, some substituents would push more electron density onto the -COO¯ group.
Would you expect trimethylacetic acid (pivalic acid or 2,2-dimethylpropanoic acid) to be a stronger or weaker acid than acetic acid? Explain your answer.
Write in your notebook, then left-click here for an explanation.
Pivalic acid is a weaker acid than acetic acid. The carbon atom has same electronegativity as other carbon atoms, and the hydrogen atoms are less electronegative than carbon. The overall effect is that a methyl group would increase the electron density on the already negative -COO¯ group. (This applies to any alkyl group, such as ethyl or propyl groups.) Hence, the methyl group’s inductive effect is in the opposite direction as the halides.
Increasing the electron density on the -COO¯ group destabilizes the conjugate base. This would lead to pivalic acid being a weaker acid compared to acetic acid. pKa of pivalic acid is 5.0.
Activity: Inductive Effect 2
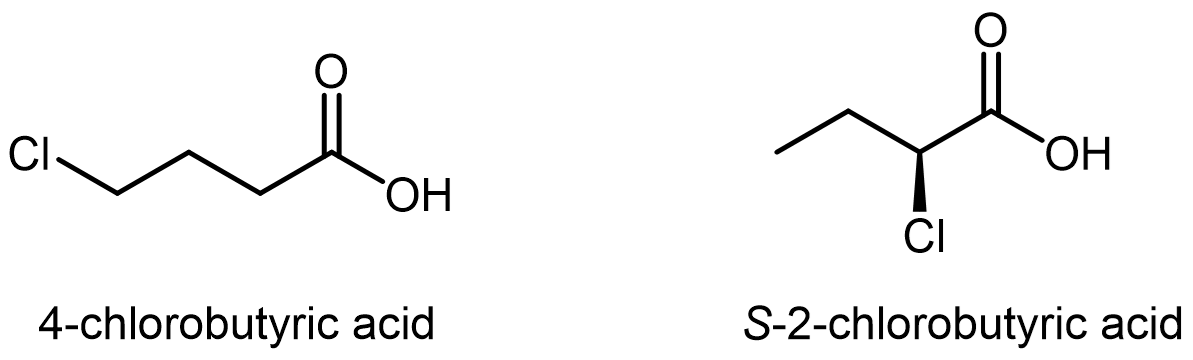
Compare the two acids below. Which one would you expect to be the stronger acid? Explain your answer.
Write in your notebook, then left-click here for an explanation.
S-2-chlorobutyric acid is the stronger acid of the two. The Cl atom is closer to the -COO¯ group (there are less C-C bonds between it and the excess electron), allowing the Cl to have a stronger inductive effect.
Looking up the pKa values, we see that 4-chlorobutyic acid has pKa = 4.5 and s-2-chlorobutyric acid has pKa = 2.9.
Oxoacids
The acidity of oxoacids, with the general formula HmXOn (with m = 1-3 and n = 1−4), depends strongly on the number of terminal oxygen atoms (oxygen atoms only bonded to the central atom X).
Because oxygen is the second most electronegative element, adding terminal oxygen atoms causes strong inductive effect, thereby increasing the strength of the acid. For example, the figure below shows how the H atoms in H2SO4 is more δ+ (more blue) than those in H2SO3, making it easier for the acid to lose the H+ in an acid-base reaction.
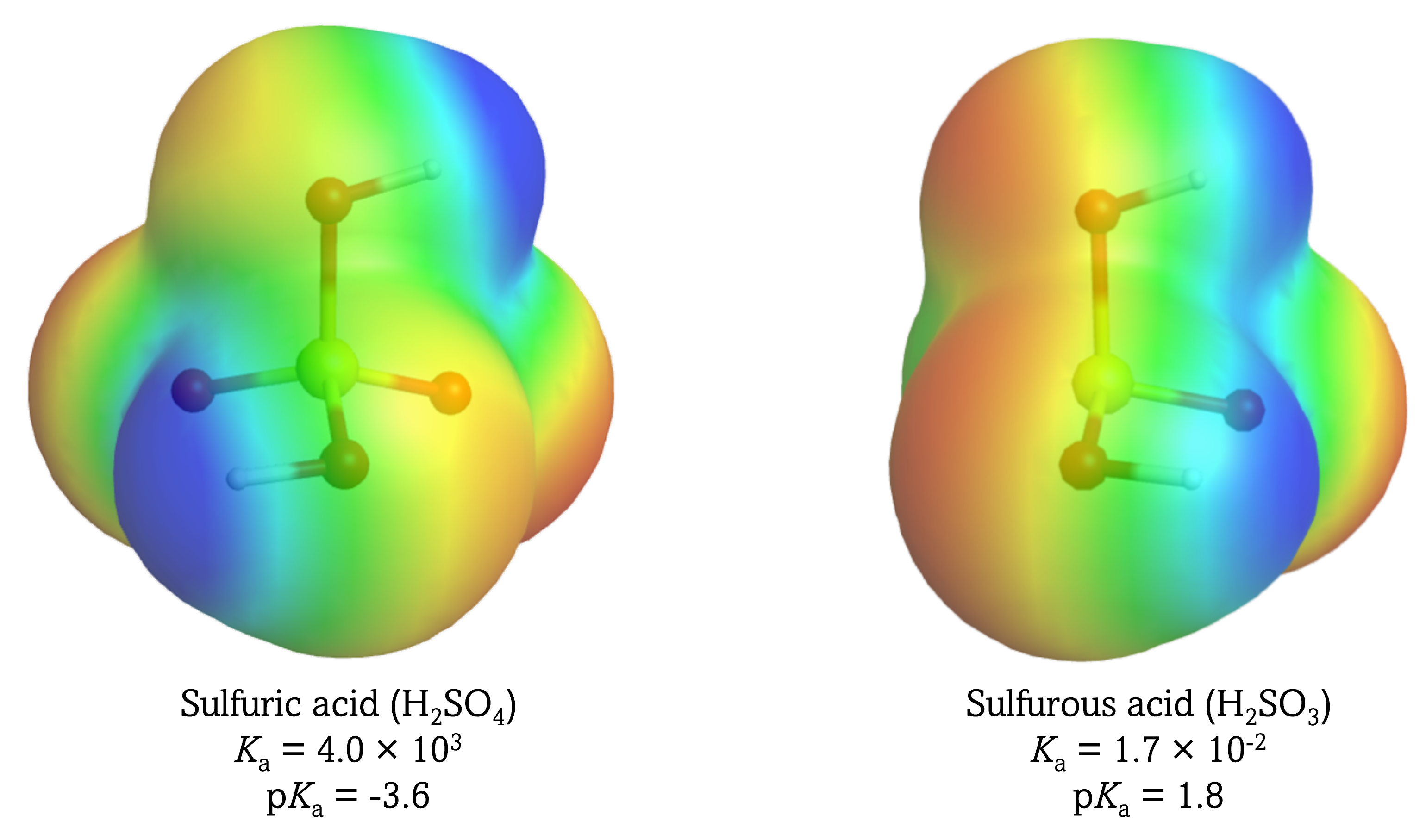
In addition, the O atoms in HSO4¯ are less δ¯ than those in HSO3¯, stabilizing the conjugate base.
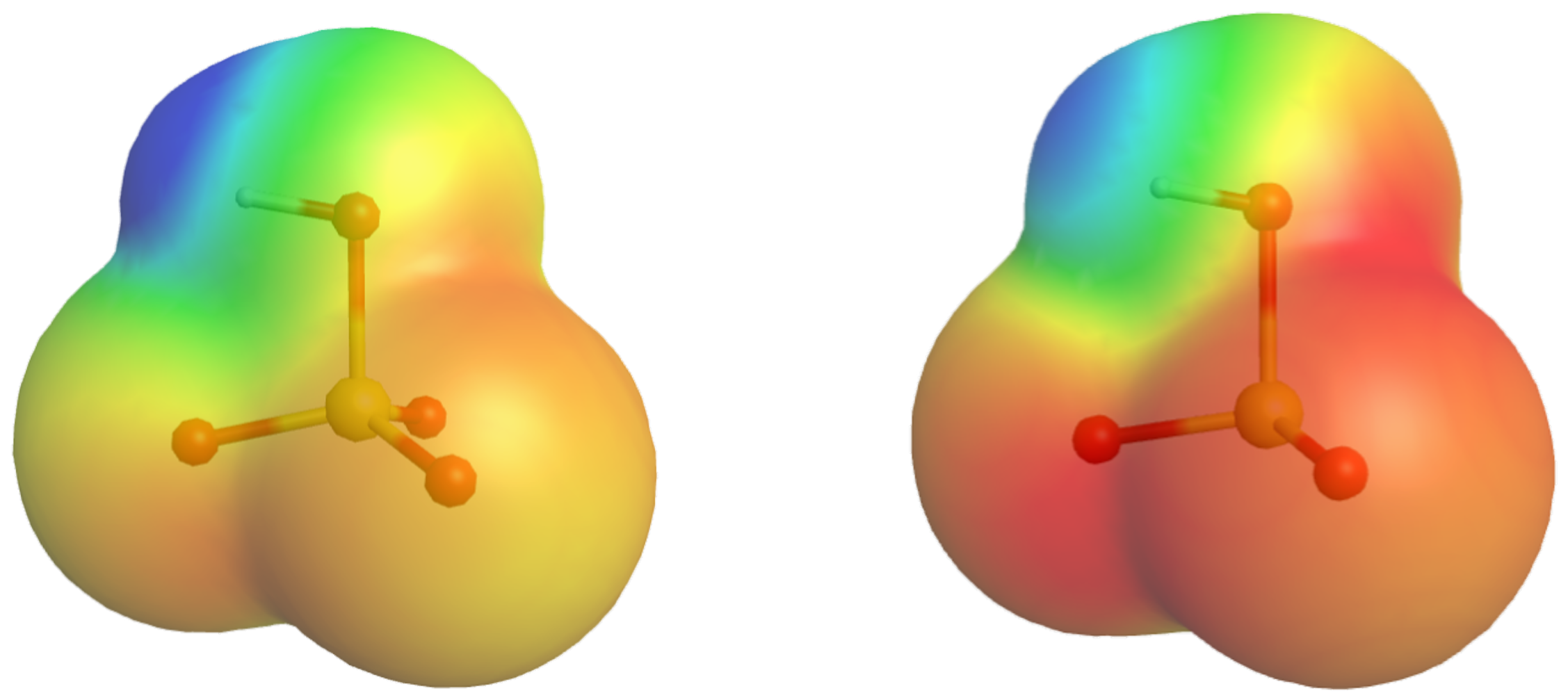
If we consider a series of oxoacids, we see that H3PO4 (one terminal O atom) is a weak acid (Ka = 7.2 × 10−3), H2SO4 (two terminal O atoms) is a very strong acid (Ka = 4.0 × 103), and HClO4 (three terminal O atoms) is even stronger (Ka = 3 × 107). This trend is explained by the number of terminal oxygen atoms, which increases steadily from P to Cl, consistent with the observed increase in acidity. In addition, the electronegativity of the central atom also increases, which further enhances the inductive effect.
Please use this form to report any inconsistencies, errors, or other things you would like to change about this page. We appreciate your comments. 🙂 (Note that we cannot answer questions via the google form. If you have a question, please post it on Piazza.)
