
Lab 3 Background
Screening Yeast Colonies
After transformation, screening for cells that have taken up the inserted DNA (often simply called an insert) are often performed. Screening is necessary because cells may or may not be transformed with all the DNA fragments and may or may not assemble the DNA fragments correctly.
The plated yeast cells grow in the unit of colonies on a plate (Fig. 3.1). Each colony is assumed to have originated from a single yeast cell (remember, yeast cells can reproduce asexually through budding). Hence, all yeast cells of a single colony contain the same genetic information. We therefore can screen each colony for its genetic makeup, in this case, whether the T7 phage genome DNA is present or absent from the yeast colony.
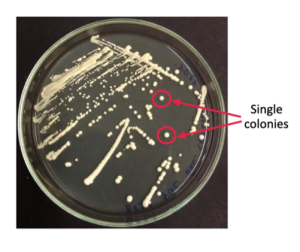
There are many ways to screen yeast colonies. The method used in this lab, which is the most common method, is the colony PCR screening method. The key steps to perform colony PCR are to design insert-specific primers to detect the presence of your insert, to set up and run a PCR reaction using the yeast cell genomic DNA and to visualize and analyze PCR result via gel electrophoresis.
The most important step of this method is primer design. Since the goal of the colony PCR is to detect the presence or absence of the insert, the primers must be designed such that only the presence of the insert can yield a band or bands. For example, to detect if fragment 1 and fragment 2 are correctly assembled, a primer set can be designed to amplify the fragment 1/fragment 2 sequence junction (Figure 3.2). If fragment 1 and 2 are correctly assembled, then a product will be yield (Figure 3.2, left panel); if fragment 1 and 2 are not assembled correctly (or if the yeast clone only has fragment 1 or fragment 2), then no band will be yielded after the PCR reaction (Figure 3.2, right panel).
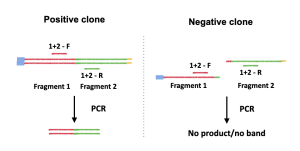
Since we transformed the yeast with multiple DNA fragments, multiple amplifications will be done simultaneously to detect the presence of all fragment junctions in the T7 genome, namely, the F1-F2 junction, the F2-F3 junction, the F3-F4 junction and the F4-F5 junction. To do this, we will use multiple primer sets in one PCR reaction, which will generate 4 bands if all the fragments are present in the yeast. Because multiple bands are generated simultaneously using multiple sets of primers, this type of PCR is also called multiplex PCR.
Multiplex PCR is different from regular PCR in that multiplex PCR contains multiple sets of primers per reaction while regular PCR has only one set of primers per reaction. While multiplex PCR is more convenient, it is not always achievable since all primers used in a multiplex PCR need to be compatible and the resulted PCR products have to be similar in length. Therefore, multiplex PCR is not often used.
Polymerase chain reaction (PCR)
PCR is a laboratory adaptation of DNA replication that uses polymerase to rapidly make millions and billions of copies of DNA. It can be used to amplify an entire DNA region, but it is more often used to amplify a segment of the DNA that the experimenter is interested in. For instance, if the experimenter is interested in studying gene A on chromosome X. Instead of amplifying the entire chromosome X, the experimenter can use PCR to amplify gene A only. In addition to making copies of DNA, PCR can be used to introduce mutations in DNA by introducing a mutation in the primers. which are short strands of DNA that are needed for PCR. Mutations can be introduced using primers because primers will become part of the newly synthesized DNA.
- Phusion Polymerase: in our laboratory, we will use Phusion polymerase, a heat-resistant enzyme that can recognize DNA and use nucleotides, the building blocks of DNA, to make copies of the DNA template. The Phusion enzyme is a fast-action enzyme that can make 1kb of DNA in about 15 seconds. To make copies, the Phusion polymerase needs a starting point, which is provided by the primers.
- Primers: primers are short pieces of single-stranded DNA designed by the experimenter that is at least partially complementary to the DNA template. By binding to the DNA template, the primers define the location of the amplification. If the experimenter wants to amplify gene A, for example, the experimenter will design two primers, one binding to the very beginning of gene A and the other binds to the end of the gene. Once the primers bind to the DNA template and act as a starting point for the Phusion polymerase, the polymerase can fill in the remaining gene sequence and make a copy of the gene. If a mutation is desired, one can design primers that are slightly different from the template DNA sequence. If the difference is not much, the primers still bind to the DNA template, and the final product, which begins with the primer sequence, will contain the mutation.
Agarose Gel Electrophoresis
Agarose gel electrophoresis is a technique that uses electrical current to separate nucleic acids based on size or shape. Agarose, a main component of agar, forms a porous gel that nucleic acids can migrate through when the current is applied. Because nucleic acids are negatively charged, they can migrate from the negatively charged side of the current to the positively charged side during the electrophoresis. This separates the nucleic acids by size, with the smallest nucleic acids migrating faster through the pores and running further down the gel.
Different concentrations of agarose are used in gels depending on the sizes of the nucleic acids: the larger the nucleic acid, the lower concentration of agarose. For technical reasons, agarose gels generally range from 0.5% to 2.5% weight by volume (W/V). In this experiment, you will run a 1% agarose gel.
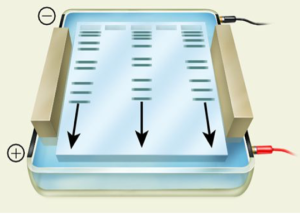
In this laboratory, your PCR DNA samples will be mixed with a 6X loading dye (1 μL of loading dye per 5 μL sample) containing tracking dyes and sucrose. While the dense sucrose helps the samples to sink to the bottom of the wells, the tracking dyes function as trackers of the sample migration during gel running.
Reference
Brunstein, J. (2013). PCR: the basics of the polymerase chain reaction. MLO Med Lab Obs, 45(4), 32, 34-35. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/23822023
Huss, P., Meger, A., Leander, M., Nishikawa, K., & Raman, S. (2021). Mapping the functional landscape of the receptor binding domain of T7 bacteriophage by deep mutational scanning. Elife, 10. doi:10.7554/eLife.63775
An enzyme which brings about the formation of a particular polymer, especially DNA or RNA.
Short pieces of single-stranded DNA designed by the experimenter that is at least partially complementary to the DNA template.