Lab 11: BACKGROUND
We have already used fluorescence spectroscopy in Lab 9 to study protein stability. Another specific application of fluorescence spectroscopy is Förster Resonance Energy Transfer (FRET). FRET results from the proximal interaction between two fluorescent molecules. A donor molecule absorbs light at a specific wavelength and is excited. Once excited, the donor transfers energy to a neighboring acceptor molecule, which then becomes excited and emits light at a specific lower energy wavelength. In order for FRET to occur, the emission spectrum of the donor must overlap with the absorption spectrum of the acceptor. In addition, the two molecules must be within a distance range of 10-100 Å.
Since the 10-100 Å range is typical for biological macromolecule interactions, FRET can be applied to measure ligand binding affinity if a protein and its ligand have appropriate fluorescent properties. In this experiment, increasing concentrations of the ligand (a fluorophore, the acceptor in our FRET experiment) are titrated into a solution of the protein (our FRET donor) resulting in a change of fluorescence emission by the fluorophore. This change in emission can be used to calculate the ligand-binding affinity (i.e. the thermodynamic binding constant, KD). This value is an equilibrium constant (given in units of Molarity) of the ratios of the concentrations of species (protein, ligand, and ligand-protein complex) in a reaction.
The accuracy range of the fluorimeter and the efficiency of the fluorophore limit FRET measurement. Generally, 1 nM to 1 μM fluorophore can be accurately detected in conventional instruments. However, in order to calculate a binding constant, the protein concentration cannot be higher than the KD. Therefore, if the binding interaction is very tight (indicated by a very low KD), the protein concentration, and in turn fluorescence, may be too low to be measured. Similarly, if one was detecting a fluorescent ligand rather than the protein, the concentrations needed to measure the high affinity will be too small to detect. Conversely, if the interaction is too weak, one might not have enough ligand to reach saturation of the protein.
For this experiment, you will calculate the binding affinity of Dansyl amide (DNSA) to HCAII using the FRET that occurs between the tryptophan residues in the active site of HCAII and the small molecule DNSA. The Trp residues in the protein absorb at 280 nm and emit at 340 nm. DNSA absorbs at 340 nm and emits light at 470 nm. Therefore, when we excite at 280 nm, we will only observe fluorescence at 470 nm if DNSA is proximal to the Trp residues of the active site. You will confirm this with controls. You will then determine the binding constant of DNSA to both mutant and wild-type HCAII.
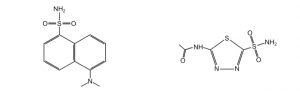
We will also measure the binding constant of acetazolamide (AZ), an inhibitor (see figure below). AZ does not fluoresce and binds very tightly to HCAII. Since this interaction would be difficult to measure directly, one can use the KD of DNSA and create a competition between DNSA and AZ to calculate the KD of AZ binding. We will also measure the affinity of DNSA and AZ to the mutant HCAII to see the effect of mutations on ligand binding.