Lab 4: BACKGROUND
Preparing for Gibson Assembly
Restriction digest
Restriction enzymes are proteins that cut dsDNA at very specific recognition sequences. The enzyme you will be using (DpnI) recognizes methylated DNA. E. coli contains enzymes that naturally methylate its DNA, so that any plasmid that originated in a bacterium will be methylated. PCR products, however, will not be methylated and so will not be digested by DpnI. This digest then allows us to eliminate the template plasmids, which are not the clones we are trying to obtain, but are still present in the PCR tubes along with our products.
The New England Biolabs website is an excellent source of information on restriction enzymes and digestions (link opens in a new tab): www.neb.com
DNA Purification
After digestion, DNA must be purified using a spin column. The spin columns contain a resin (white) that will bind to DNA, while other components will flow through into the collection tube.

For more molecular details on spin columns, please see the lab manual appendix: Appendix 3: Additional laboratory details
Spectrophotometric Determination of DNA Concentration and Purity
Biological molecules absorb light of different frequencies in a characteristic manner, and we can take advantage of this using spectrophotometry. In general, DNA exhibits maximum absorption of light at 260 nm. In this lab, we will use a spectrophotometer called a NanoDrop to measure the absorbance at 260 nm, or A260, of your purified DNA samples. The NanoDrop reads 2 μL of sample. The inset image below shows the pedestal (within the black circle) where the 2 μL of sample needs to be placed.

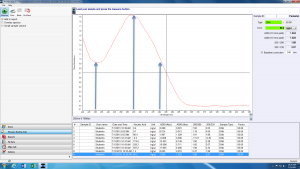
Look at the sample NanoDrop absorbance plot below. The arrow in the center highlights the point of most interest to us: the absorbance at 260 nm. The NanoDrop uses this number to calculate the concentration of DNA (96.2 ng/uL in this example).
The absorbance at 230 nm (left arrow) should be low. If it is high, it may indicate a small molecule contaminant, such as EDTA or carbohydrates. Expected A260/A230 values for pure DNA are in the range of 2.0-2.2. If the ratio is appreciably lower, it may indicate contaminants that absorb at 230 nm.
The absorbance at 280 nm (right arrow) should also be low. The expected A260/A280 value for pure DNA is ~1.8. If the ratio is appreciably lower, it may indicate the presence of protein in your sample.
Gibson Assembly
Developed in 2009, Gibson assembly is a modern approach for joining multiple DNA fragments into a desired clone. In our case, we will be assembling the HCAII gene into the protein expression vector pETblue2. However, Gibson assembly can also be used to join up to ten DNA fragments.
If you are interested, here is the original paper describing this method (pdf opens in a new tab): 2009 Gibson cloning reference
Gibson assembly requires that DNA fragments contain ~30 bp of overlapping sequence identity. Typically, this sequence overlap is generated via PCR – insert and vector are amplified via PCR using primers that create sequence overlap. PCR products can then be digested with the enzyme DpnI to eliminate template DNA.
Purified DNA fragments with overlapping sequence are then joined in a single, isothermal reaction. The reaction mixture contains three enzymes, in addition to a number of buffer components to optimize function of each enzyme. Below are the steps in Gibson assembly, with the required enzymes underlined:
- Exonuclease chews back each strand of DNA from the 5’ end. This creates single-stranded, sticky overhangs. Overlapping sequences can anneal together.
- DNA polymerase adds nucleotides to fill in any gaps.
- DNA ligase covalently links the annealed DNA fragments, thereby creating a completely double-stranded circular piece of DNA: your clone!
Transformation
When bacterial cells take up DNA they are said to undergo “transformation.” Host bacterial cells must be in a state called “competence” which is achieved by treating healthy, mid-log-phase cells with a solution of ice-cold calcium chloride and then freezing them. The cells are then thawed, mixed with the DNA that you wish to transfer, and subjected to a brief pulse of heat, called a “heat shock”. This process makes the bacterial cell membrane porous and creates a gradient such that DNA can pass through the membrane and into the cell.
Competent cells are very fragile and must be treated with care: keep on ice and mix very gently!! The timing and temperature of the heat shock step must be precise; if kept too long at the high temperature, the cells will die. Furthermore, careful aseptic technique must be used throughout, because any contaminating bacterial cells may grow and compete with desired cells.
After the recombinant DNA is transferred to the competent host cells, the cells are incubated in rich medium to allow them to recover from the stress of the heat shock, as well as to begin growing and replicating the newly transferred DNA. Following the recovery period, aliquots of the culture are plated on a selective medium, so that only clones that received the plasmid will grow. The resulting bacterial colonies are then screened to locate those that have the desired characteristics.
For additional information about transformation, please see the appendix: Appendix 3: Additional laboratory details
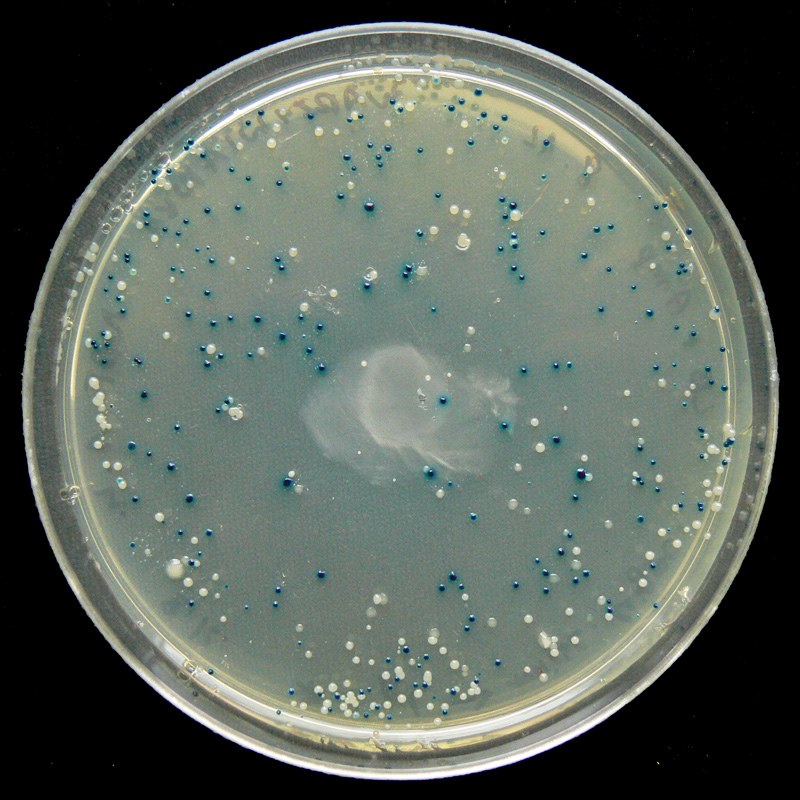
Blue/White Screening
Some vectors, such as pETblue2, allow for rapid screening of potential clones using the blue/white method. Screening is necessary because transformed colonies from a cloning reaction may or may not contain the desired clone – screening methods allow you to confirm the presence of your insert.
In the blue/white screening method, the gene of interest (HCAII in our case) is inserted into the vector such that it will disrupt the lacZ gene. You can see the lacZ gene in pETblue2 in the vector map here: pETblue2 vector map
When looking at the vector map, note that inserting HCAII into the Multiple Cloning Site will disrupt lacZ. The lacZ gene encodes for the protein β-galactosidase, which functions to cleave lactose. When plated on LB-agar plates containing X-gal, which is an analog of lactose, cells with in tact β-galactosidase will turn blue. However, in cases where lacZ has been disrupted by a cloned insert, the colonies will remain white. Therefore, after plating on LB-Amp-Xgal plates, you will be able to count the number of blue (negative) and white (positive) clones. You will then pick several white colonies to further screen.